Temel olarak nöromusküler hastalıkları 3 gruba ayırabiliriz.
- Progresif Musküler Distrofiler (Duchenne, Becker, Emery-Dreifuss, Fasio-Scapulohumeral ,Limple-Girdle ,Distal ,Oküler ve Konjenital MD)
- Herediter MD (Tip 1 ,2,3,4 SMA)
- Nöromusküler Kavşak Hastalıkları (Myastenia Gravis ,Lambert-Eaton myastenik sendromu ,Konjenital myastenik sendromu )
Bugün burada bu hastalıkların bazılarından bahsedip en azından birazcık konu hakkında bilgi sahibi olacağız.O zaman öncelikle MD ne demek onu bir açıklayalım.
Musküler distrofi; alt motor nöronda yapısal bir anormallik olmaksızın iskelet kaslarının dejenerasyonu ile karakterize olan kaslarda güçsüzlük ve atrofiye (erime) sebep olan bir hastalıktır.Progresif MD ‘ler asıl problem inervasyonda ve terminal sinir sonlarında değil kasın fibrillerindedir bizzat. MD’ler de çizgili kas liflerinde bağ dokusunda artış ,kas liflerinde atrofi ve pseudohipertrofi ,kalsiyum birikmesine bağlı olarak hiperkontrakte lifler ,normalde periferde olmasını gereken çekirdeğin merkeze doğru kaymış olması (internal nucleus durumu ) ve dejenerasyon görülür.
Duchenne MD
En sık görülen MD çeşididir.X’e bağlı resesif geçişlidir.DMD geni 21. X kromozomuna ait ve distrofin (hızlı kasılan tip 2b fonksiyonunda bozukluk) proteinin kodlanmasından sorumludur.
DMD tanı kriterleri;
- Semptomların 2 yaş öncesinde görülmesi
- Simetrik progresif kas güçsüzlüğü (proksimal kasların önce tutulması ,alt ekstremitelerin önce etkilenmesi ve baldırda pesudohipertrofi durumu)
- 13 yaş öncesinde tekerlekli sandalyeye ( TS ) bağlanma
- Distrofin geninde meydana gelen delesyon
- Fasikülasyon ve duyu kaybının olmaması
Genetik bir neden yoksa genelde 5 yaşında teşhis konulur ama semptomlar 2,5 yaşında başlar.İlk gerginlik gastro-soleus(baldır kaslarımız) ve TFL kasında meydana gelir.Hastaların nerdeyse %50 si 18 aya kadar yürüyemez.
Peki DMD’de klinikte neler gözümüze çarpar ?
- Sık sık düşmeler ve sakarlıklar göze çarpar.
- Waddling gait denilen ördekvari bir yürüyüş
- Gower’s pozitiftir bu hasta grubunda.
- Baldırda pesudohipertrofi
- Merdiven inme-çıkmada ve özellikle yerden kalkma da problem yaşarlar.
- Yürüyüşlerine gelecek olursak; destek yüzeyleri artmıştır.Lateral(yana) gövde salınımları artmıştır.Parmak ucunda yürüme çok bariz gözümüze çarpar.Omuzlarda retraksiyon(geriye çekilme) ve kol salınımlarında azalma durumu göze çarpar.
- Zamanla DTR (derin tendon refleksleri ) azalır hatta kaybolur.
- 10 yaşın altında kardiyomiyopati denilen kalp rahatsızlığı görülür.
- Mental reterdasyon görülebilir.Özellikle yavaş okuma, kelime bulmada güçlük ve bellek zayıflığı da olabilir.
Becker MD
Sadece erkeklerde görülen bir hastalıktır.DMD ‘nin yavaş ilerleyen formudur .Semptomları 11 yaş civarında başlar ve ambulasyon kaybı 27 yaşında olur.Özellikle peroneal ve tibialis anterior kasının tutulumu tipiktir bu hastalıkta.DMD ‘den farklı olarak kalp tutulumu daha yoğundur.Baldırda pseudohipertrofi burada da mevcuttur.Kardiyak miyopatiler yaşam sürelerini kısıtlayan en önemli faktördür ve 40’lı yaşlarda ölürler.
Fasio-Scapulohumeral MD
Otozomal dominant geçişli olan bir hastalıktır.Temel reis görünüme sahiptir hasta grubudur.Yüz, skapular, omuz, bacak, kalça kuşağı kaslarının sıklıkla asimetrik etkilendiği, yavaş ilerleyen kas güçsüzlüğü ile karakterizedir.Çizgisiz yüz, transvers gülümseme ,tapir dudağı ve konuşma bozukluğu gibi özelliklere de sahiptir bu hasta grubu.Hastalıkta ilk önce fasial kaslar tutulur. Gözleri sıkı kapatmakta, gülmekte, ıslık çalmakta yetersizlik, dudaklarda somurtkan görünüm, donuk yüz ifadesi, ağız köşelerinde çökme izlenir.
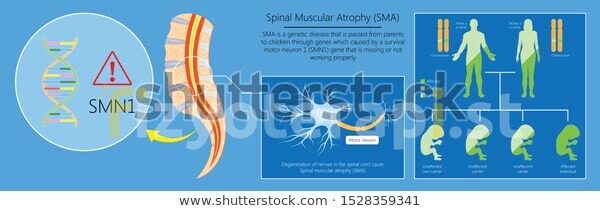
Nöromusküler Hastalıklar ve Spinal Musküler atrofi (SMA)
SPİNAL MUSKÜLER ATROFİ (SMA)
İskelet kaslarında atrofiye ve tüm vücutta genel bir zayıflığa yol açan ön boynuz motor nöronlarda dejenerasyonla karakterize nörodejeneratif bir hastalıktır.Çocukluk çağında DMD hastalığından sonra en sık görülen nöromusküler hastalık grubudur.
- Akut infantil…….Tip 1 SMA
- Kronik………Tip 2 SMA
- Juvenil……….Tip 3 SMA
- Yetişkin………Tip 4 SMA
Atrofinin nedeni ;kaslara normal fonksiyon görebilmeleri için gerekli olan elektriksel ve kimyasal sinyallerin doğru bir şekilde dağıtılamamasıdır. Spinal kordun ön köklerinde incelme varken posterior duyu kökleri korunuyor.DMD ‘den en büyük farkı ise atrofik gruplar arasında fibrozis olmaması durumudur. Otozomal resesif bir genetik bozukluktur ve 5. kromozomda yer alan (5q13) SMN1 geninde mutasyon meydana gelmiştir.SMA oluşumunda rol oynayan bir diğer gen de NAIP dir.SMN ,RNA’nın protein üretimine katılıyorken ,NAIP ise kasları uyaran sinir hücrelerinin ölümünü engeller.
SMA tip 1 (Werding-Hoffman Hastalığı)
- Doğumun ilk 6 ayında görülen SMA grubunun en ağır tipi olan hasta grubudur.
- Kas fasikülasyonları sıklıkla görülür.
- Primer nedeni, boynuz motor nöronların progresif kaybına bağlı olarak görülen kas zayıflığıdır.
- İlk 6 ay içerisinde meme emmede güçlük, disfaji ve solunum güçlüğü görülebilir.
- 6.ayda motor hareketlerin gecikmesi dikkat çeker.
- Respiratuar kapasite azalır dolayısıyla hemen yorulurlar.
- Gövde ve ekstremiteleri de içine alan şiddetli zayıflık ve hipotoni dikkat çeker.
- Hiçbir zaman baş kontrolü kazanamazlar ve desteksiz oturamazlar.
- Bacaklarını kaldıramazlar.Kalçada abdüksiyon, eksternal rotasyon (dışa doğru) ve dizlerde fleksiyon (bükülme)ile karakterize kurbağa pozisyonu tipiktir burada .
- İnterkostal kaslar ciddi olarak etkilenmiştir bu nedenle diyafragmatik solunum gözlenir.
- Göğüste inspiasyon esnasında abdomenin genişlemesi ve kostaların içeri çekilmesi ile Çan şeklinde bir görünüm göze çarpar.
- Bulbar kaslarındaki zayıflıktan dolayı emme ve yutma zorlaşır.Ağlama zayıf ve güçsüzdür.
- Duyu bozukluğu yoktur DTR’ler alınamaz.
- Vakaların büyük bir kısmı tekrarlayan respiratuar enfeksiyonlar ve beslenme sırasında yiyeceklerin aspirasyonu nedeniyle 2 yaşın altında kaybedilirler.
SMA Tip 2
- 7-18 ay arası başlangıç gösterir.Bu hastalar desteksiz oturmayı becerebilirler ve bazıları emekleyebilir ve hatta ayakta durmayı başarabilirler, fakat desteksiz ayakta durma ve yürümeyi başaramazlar.
- DTR ‘ler kaybolmuştur ve üst ekstremitelerde tremor yaygındır.
- Solunum fonksiyonlarını olumsuz etkileyen ve çok hızlı ilerleyen skolyoz durumu mevcut olabilir.
SMA tip 3(Juvenil Kugelberg-Welander)
- SMA ‘nın en hafif formudur 2 yaşından sonra başlar.Genellikle ilk belirti yürüme zorluğudur.
- Bulbar semptomlar yoktur.
- Kalça ve omuz kuşağı kas gruplarında zayıflık
- Spinal deformiteler olabilir.
- Gower’s pozitiftir ve serratus anterior kasının etkilenimine bağlı olarak scapula alata mevcuttur.
- 30 yaş civarında genelde hastalar tekerlekli sandalye kullanırlar.
- Yaşam süreleri normal sayılabilir.
SMA Tip 4
Hastaların yetişkinlik döneminde bağımsız yürümeye devam edebildiği ,solunum ve yutma fonksiyonlarının etkilenmediği ılımlı bir SMA formudur.
Peki bir tedavi methodu mevcut mudur ?
Aslında bilinen kesin bir tedavi yoktur.Özellikle akciğerde yaşanan problemler Tip 1 ve 2 hastalarında temel morbidite ve mortalite sebebidir.
Peki SMA ‘da fizyoterapinin amacı nedir?
Kas ve eklemeleri koruyarak oluşabilecek limitasyon ve deformitelerin önüne geçebilmek, solunum fonksiyonlarını en iyi düzeyde tutmak ve akciğer komplikasyonlarından korumak ,hastaların yaşam kalitelerini ve sosyal katılımlarını artırabilmektir.

{kind=link}